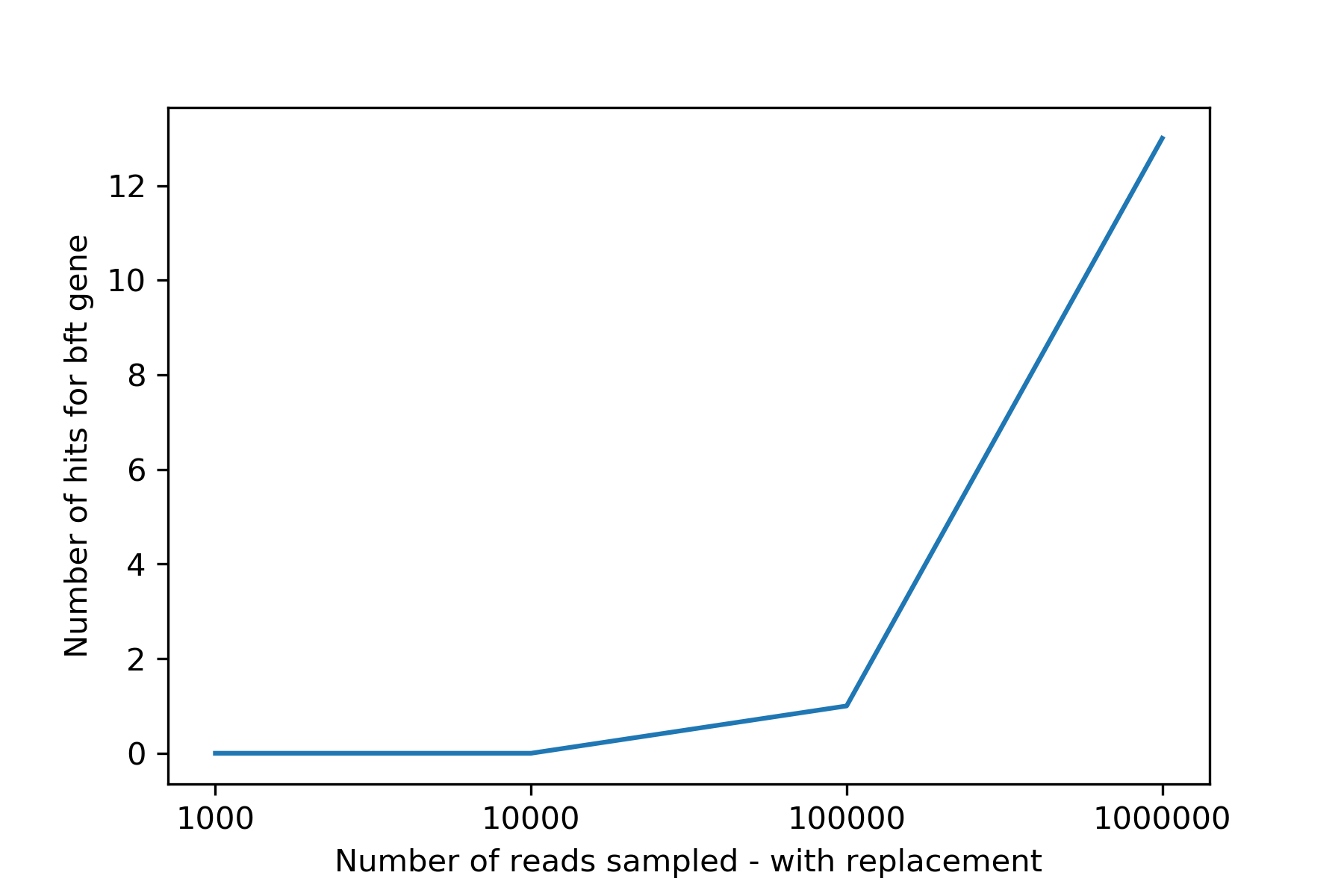
seqDepth
Question:: Is ability to detect "a gene" a function of seqencing depth?
Here you can find approaches to:
i) Sampling a fasta file randomly with replacement
ii) Example of creating a HMM profile for gene
iii) Search genes predicted from a genome against HMM profiles
##### Tools/Program used:: prodigal
Prodigal (Prokaryotic Dynamic Programming Genefinding Algorithm) is a microbial (bacterial and archaeal) is a gene/ORF finding program.
Hmmer provides tools for making probabilistic models of protein and DNA sequence domain families –called profile hidden Markov models, profile HMMs, or just profiles – and for using these profiles to annotate new sequences, to search sequence databases for additional homologs, and to make deep multiple sequence alignments. Steps: 1) Multiple sequence alignment of a homologous family of protein 2) Making HMM models using the aligned output 3) Use the constructed HMM profile to annotate other sequence/aa.
MUSCLE (MUltiple Sequence Comparison by Log-Expectatio) is multiple alignment programs.
##############################
This code allows to randomly sample a large fasta file at n number of times with replacement. Once sampled, then gene is predicted for each sub-sampled fasta files using prodigal. Predicted gene file is searched for the gene of interest using HMM profile in hmmer. In order to run hmmer search, at first protein sequnces for a gene of interest is downloaded from uniport, aligned with muscle, and HMM profile is created with hmmbuild function from Hmmer. The built HMM profile is then used to search gene of interest in the sub-sampled fasta files.
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
"""
Created on Wed Jun 10 17:03:23 2020
@author: ravinpoudel
"""
import os
import subprocess
import pandas as pd
import matplotlib.pyplot as plt
from Bio import SeqIO
from random import choices
from random import seed
# input fasta file
fasta = "MSM9VZPH_contigs.fna"
def rsamplefasta(fasta, n, nseed):
"""Randomly sample n number of reads - with replacement"""
# fname to write as an output filename
fname = os.path.splitext(fasta)[0] +"_"+ str(n) +".fasta"
# index fasta file, allows for fast access
record_dict = SeqIO.index(fasta, "fasta")
# create a list of sequence header
seqheader = []
for file in record_dict.keys():
seqheader.append(file)
# randomly sequence for seqheader for 'n' times
seed(nseed)
sub_header = choices(seqheader, k=n)
# retrive reads/seqrecord from fasta file, based on header in seqheader list
sample_fasta = []
for header in sub_header:
sample_fasta.append(record_dict[header])
# write down as a fasta file
SeqIO.write(sample_fasta, fname , "fasta")
def rangesampler_fasta(fasta):
"""Randomly sample [1000, 10000, 100000, 1000000] reads - with replacement"""
# sample range
samplerange = [1000, 10000, 100000, 1000000]
# random seed
nseed = 12121232
# sample randomly n times and save as a fasta file
for i in samplerange:
print("Ranomly Sampling", i, "sequences")
rsamplefasta(fasta, i, nseed)
#rsamplefasta(fasta, 20, nseed)
# Randomly sample reads from a given fasta file @ n = 1000, 10000, 100000, 1000000
rangesampler_fasta(fasta)
# make a gene prediction using prodigal , for each n sampled fasta files
subprocess.call("./prodigal.sh")
## Run hmmer search - with gene of interest - eg. bft here.
# basic order: gene faa > align with mafft > hmm profile > hmmsearch
subprocess.call("./alignthenHmmer.sh")
## Now count the number of hits for each file
subprocess.call("./counthits.sh")
### count the number of matches
lcount = pd.read_csv("countline.txt", header=None, sep=" ", names=['Filename','nhits'])
lcount['nreads'] = lcount.Filename.str.rsplit("_", n=1, expand=True)[1].str.rsplit(".bft.txt", expand=True)[0].to_list()
### Create a plot
plt.plot(lcount.nreads.to_list(), lcount.nhits.to_list())
plt.ylabel('Number of hits for bft gene')
plt.xlabel("Number of reads sampled - with replacement")
#plt.grid(True)
plt.savefig('bft.png', dpi=300)
plt.show()